












Case of the Week (COW)

Current COW
COW Winners
Recently Posted
Current COW
COW Winners
Recently Posted
Current Case of the Week (COW)

Current Case of the Week (COW)
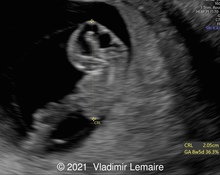
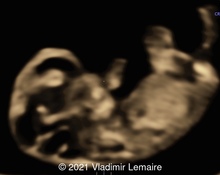
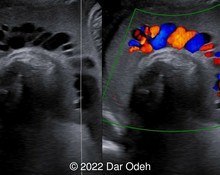
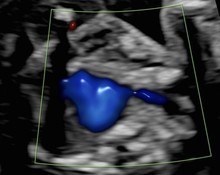
A 36-year-old pregnant woman (G2P1) was referred at 18 weeks and 4 days gestation to our tertiary center for sonographic evaluation due to the suspicion of various fetal anomalies. Test
Submit Your Answer
To join the community and participate in solving current cases, simply click the "view current case" button below. This will take you to the case summary page with full details and additional media. To participate, you will first need to create an account or sign-in. You can then submit your answers.
You can only submit your answers once, but you get three answers. The correct answer is revealed at the end of the posting period.
Physicians
Previous Winners
-

Javier Cortejoso, Spain
Cases Solved: 2 -
Vladimir Lemaire, United States
Cases Solved: 2 -
Andrii Averianov, Ukraine
Cases Solved: 2 -
Alexandr Krasnov, Ukraine
Cases Solved: 2 -
Mayank Chowdhury, India
Cases Solved: 2
First-Time Winners
-

Gulten Rafibeyli, Azerbaijan
Cases Solved: 2 -
Ayten Sadigova, Azerbaijan
Cases Solved: 2 -
Mert Eyupoglu, Turkey
Cases Solved: 2 -
Gulsum Mammadova, Azerbaijan
Cases Solved: 2 -
Vu The Anh, Viet Nam
Cases Solved: 2
Sonographers
Previous Winners
-

Padman KG, United Kingdom
Cases Solved: 2 -
Eti Zetounie, Israel
Cases Solved: 2 -
Anette Beverdam, Netherlands
Cases Solved: 2 -
Dianna Heidinger, United States
Cases Solved: 1 -
Kimberly Delaney, United States
Cases Solved: 1
First-Time Winners
-

Sonio Sonio, France
Cases Solved: 1 -
Amy Scheible, United States
Cases Solved: 1 -
Philippe Viossat, Antarctica
Cases Solved: 1
Contributors
Top Contributors
-
Javier Cortejoso, Spain
Article & Case Contributions: 3 -
Victoria Giang, Viet Nam
Article & Case Contributions: 1 -
Fahimeh Azizinik, Iran, Islamic Republic of
Article & Case Contributions: 1 -
Jeanine Coetzer, Australia
Article & Case Contributions: 1 -
Vladimir Lemaire, United States
Article & Case Contributions: 1
Previous winners: Users who have been recognized on a "Top Winners list" in years past; First-time winners: Users who have not yet been on the annual "Top Winners list." The Sonographers category also incorporates "other" job titles.
News & Notes
Dear Esteemed Users of TheFetus.net,
We had 22 cases of the week this year with unique and complex conditions that varied from Torcular Herophili Thrombosis to Prune Belly Syndrome to Sternoschisis. We want to hear which one was your favorite!
Your favorite case may be the one with the most demonstrative images and videos, a particularly well-written and informative discussion, or an unusual and unique disease process. To vote for your favorite case, log into… read the full entry
TheFetus.net
© 2014 - 2026 TheFetus.net